
Overview
Previously we looked at some of the basic principles of nervous system function and how chemicals from several pesticide classes disrupt normal function. This time we will look in detail about what we can expect for a dose-response characterization of acetylcholine pathway insecticides based upon their mode of action and properties of the nervous system. This will get a little more technical than usual. The casual reader may want to skim over the equations but think about the explanations.
Several families of pesticides function by disrupting the synaptic acetylcholine pathway. Organophosphate pesticides and the carbamates block the enzyme acetylcholinesterase (AChE) such that the naturally released neurotransmitter, acetylcholine (ACh), is not broken down and recycled. Instead ACh builds up in the synaptic junction and over-stimulates the acetylcholine receptors (AChR) on the post-synaptic membrane.
The neonicotinoids act directly by bonding strongly to the nicotinic acetylcholine receptors (nAChR) in a manner that holds open the receptor ion channel.
Both classes of chemicals, the AChE inhibitors and the nAChR agonists, produce excessive numbers of activated acetylcholine receptors on the post synaptic membrane, which gives rise to a reduction in the post synaptic resting potential and a propensity to generate action potentials in the post synaptic neuron. Acute poisoning occurs when the general level of neural stimulation is sufficient to disrupt the normal physiological processes required to sustain life. Clinically, insects and animals poisoned with either class of chemicals are seen to lose muscular control, exhibit uncontrolled twitching, eventual paralysis, and death.
We will begin by considering a single synapse and come up with a relationship for the post synaptic stimulation as a function of the fractional lethal chemical level. We will also consider implications of acetylcholinesterase disruption for an entire neural network. Finally we will seek to understand the environmental implications of threshold versus non-threshold action with these chemicals.
Synaptic electro-chemical function
The nervous system is governed by neuronal generated “action potentials”, rapid electrical potential changes that travel rapidly along the neural axons and terminate in the branching tree of dendrites at synapses where they can cause the release of neurotransmitter into the synaptic cleft. The neurotransmitters rapidly diffuse across the synaptic junction and attach to receptors on the post-synaptic membrane. These transiently bound receptors change the permeability of the membrane and allow ion currents to flow across the membrane, thus altering the local cellular electrical potential in the post-synaptic neuron.
Action potentials are fast transients that last 1-3 milliseconds. Diffusion time of ACh across the junction is faster, measured in microseconds, and the decay of synaptic free-circulating ACh is normally around one millisecond. The time response of the excitatory post-synaptic potential is slightly slower, typically lasting several to 10’s of milliseconds.(1) This allows the post-synaptic neuron to be the summing junction from many synaptic inputs, doing some kind of dynamic averaging that determines whether or not the downstream neuron will produce its own action potential. We argue that changing the decay time of ACh in the synaptic junction relates directly to stimulation that will produce an action potential in the downstream neuron. Roughly, doubling the decay time is likely to double the likelihood of the downstream neuron generating its own action potential because the amount of post-synaptic charge transfer will be proportional to the length of time ACh holds open the AChR receptors, and this open time is within the typical averaging period of the neuron.
Acetylcholinesterase Inhibitors – Consider a single synapse
A normal stimulus, 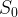
[1]

Where 

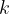

The primary way acetylcholinesterase inhibitors act is by reducing the number of AChE molecules available to catalyze the destruction of ACh in the synaptic junction. It is reasonable to expect that decreasing the number of available AChE molecules will proportionally increase the time it takes for ACh molecules to be degraded. If we assume that a fraction, 

[2]
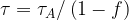
For a single action potential neither 
[3]

As 

[4]

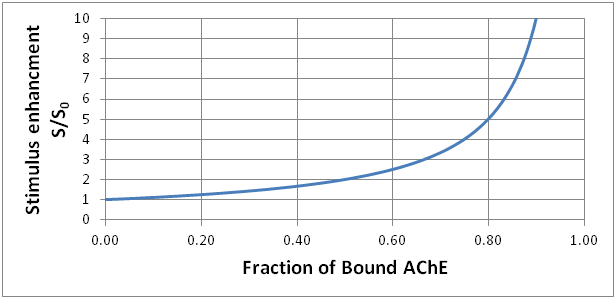
The graph shows what happens as the fraction of bound AChE increases. The stimulus enhancement rapidly gets large as most of the AChE becomes unavailable to catalyze the destruction of ACh.
With a little algebra you can show that the fraction of excess stimulation at the sub lethal limit compared to lethal over-stimulation can be expressed as
[5]

Where 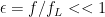
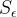

Example: If the lethal stimulus level is five times the normal background level of neuronal activity, then 80% of the AChE must be bound. If we ask what happens with an exposure that is 10% of the lethal level, (8% of AChE bound) then the increase in simulation is only 1.6% of the increase needed for lethality. In the residual limit, the stimulus increase is less-than-linear with exposure, with this “safe residual” effect strongest for chemicals where
Studies with fish have shown that AChE inhibition levels need to be 60% to 90% (2), depending upon the chemical and species, to be lethal. This is more or less in accord with this model where lethality requires most of the AChE receptors to be out of commission, and would suggest that toxicity suppression for residual levels would be significant for these chemicals.
Acetylcholine Dynamics – Consider the complete network
Now we will take the nervous system as an ensemble of neurons with average properties. Specifically we are interested in the acetylcholine pathway, so we define several global average quantities and relationships between them. Acetylcholine activates receptor sites on the post synaptic membrane that stimulate the post synaptic neuron. We can express this globally averaged stimulus, 
[6]

where 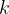

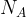
[7]

Where 


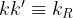
[8]

A solution of the differential equation is
[9]

Where the growth rate,
[10]

The growth rate,
[11]

Hence, under normal conditions, the concentration of AChE must be sufficient to prevent runaway growth of the ACh concentration due to ACh’s ability to generally stimulate the neural network. Here we are not considering the many other neurotransmitters, both agonists and inhibitors, that are included in the network, nor are we considering external inputs. However, conditions that place the entire network in a rough dynamic balance enhance the network’s ability to involve multiple neurons for information processing. Hence, one might suspect that the inequality [11] is only weakly maintained, at least in some portions of the neural network since this would lead to a network that would more optimal for information processing .
The complete network with AChE Inhibitors
Now let us consider what happens when we add AChE inhibitors and to this picture. The effect of chemical AChE inhibition will be to reduce the natural concentration AChE, 
[12] 
where
[13]

One might consider this the threshold level at which AChE pesticides produce a lethal effect.
In the medical literature on OP poisoning, one comes across the notion of “cholinergic crisis” which suggests such a threshold-like condition (3). Although experimentally it is found that relatively large fractions of the AChE must be inhibited to cause lethality, this network effect may play the role of the coup de grâce at the entire organism level.
Acetylcholine Receptor Agonists – Neonicotinoids
The acetylcholine receptor agonists such as the neonicotinoids will directly stimulate the post synaptic neuron. We can write the postsynaptic stimulation, 
[14]

Where 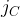
No obvious threshold mechanisms are present for this class of chemical. Instead, the excess stimulation is directly proportional to the amount of bound receptors, which is itself proportional to insecticide dose. If we go through the exercise like we did for equation [5] we discover that in the residual limit where 
[15]

showing the stimulus is proportional to the residual dose.
Metabolic load for residual levels of these chemicals
Any depolarization of the post-synaptic neuron must be eventually be rectified by metabolic processes that pump ions uphill against the gradient to return the neuron to its normal resting potential. Chemicals that increase the post-synaptic stimulation beyond the natural level will require proportionately more metabolic effort to return the neuron to its resting potential. For the AChE inhibitors the excess stimulation is only operative when the synapse is stimulated by the action potential and ACh is present. If we wish to find an averaged excess stimulation of the post synaptic neuron, we need to multiply the instantaneous excess stimulation by the synaptic duty cycle, 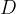
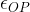
[16] 
For the neonicotinoids, the stimulation is constant, with duty cycle equal to one when doing the time averaging.
[17]

If we assume that chemical pesticides are applied in the field at rates that are designed to produce a lethal effect in target organisms, then we can compare the relative effects of residual levels of the chemicals 
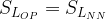


[18]

The above comparison suggests that for similar residual levels of the two classes of chemicals, the neonicotinoids will produce a much larger average post-synaptic stimulation. We can make estimates for the synaptic duty cycle based upon observed average firing frequency, ~1 Hz, and typical action potential duration, ~2 ms. If we assume the threshold term 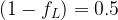
Time Cumulative Effects
The time history of the the movement of the pesticide from its initial application, its interaction with target or non-target organisms, and its eventual dilution and degradation can have dramatic consequences in terms of both acute initial toxic effect and latent residual toxic effect.(5) An effective and safe pesticide should strongly attack the target organism yet remain benign to similar species that are not the target organisms. The best way to achieve a strong differentiation from initial application compared to residual pollutant is to use chemicals that have all of the following properties:
- Rapidly degrade in the environment.
- Rapidly disassociate at targeted biological binding sites.
- Have a strong threshold action.
Lets look at these in turn. Persistent chemical pollutants have been the bane of the pesticide industry since DDT. None of the acetylcholine path insecticides are as bad as the organochlorines, but there is still quite a difference between members of this group. The neonicotinoids are said to have around a 1 year soil life, but experience suggests that to be an optimistic number. Where the chemicals have been used for many years, the contamination levels continue to increase. Since the neonicotinoids are water soluble, this suggests that what may appear as degradation is merely dilution and migration. Instead of the chemical disappearing, we find contamination far from the source of the application. (6,7,8) Chemicals that are persistent in the environment long after the crop is harvested and target insects are gone can only have deleterious consequences for unintended organisms. The severity of the consequences depends on the final two properties.
Insecticide chemicals that bind to targeted receptors can have a wide range of receptor affinity and binding strength. Chemicals that bind transiently (like the ACh molecule itself to AChRs) will remain in quasi chemical equilibrium with the extracellular fluid and will bind to target molecules at a rate that is proportional to the concentration of the chemical. However, some insecticide chemicals are designed to bind tenaciously to the desired receptor sites. In these cases, the molecules will become trapped at the target site even after most of the chemical has been rid from the organism’s body by metabolic processes. In cases with very strong target binding, one can expect accumulation over time of molecules at the target sites as long as there is any continuing exposure to the chemical. How serious a problem this will be for non-target organisms depends on the last property, whether the chemical works with a threshold action or not.
Properties of Pesticide Classes
Chemical neuro-toxic pesticides have been widely used for more than 70 years. During that time several families of chemicals have been developed to target specific neurological receptors. The chart below lists several of these classes, includes a common example or two from each class and shows typical properties of these chemicals.
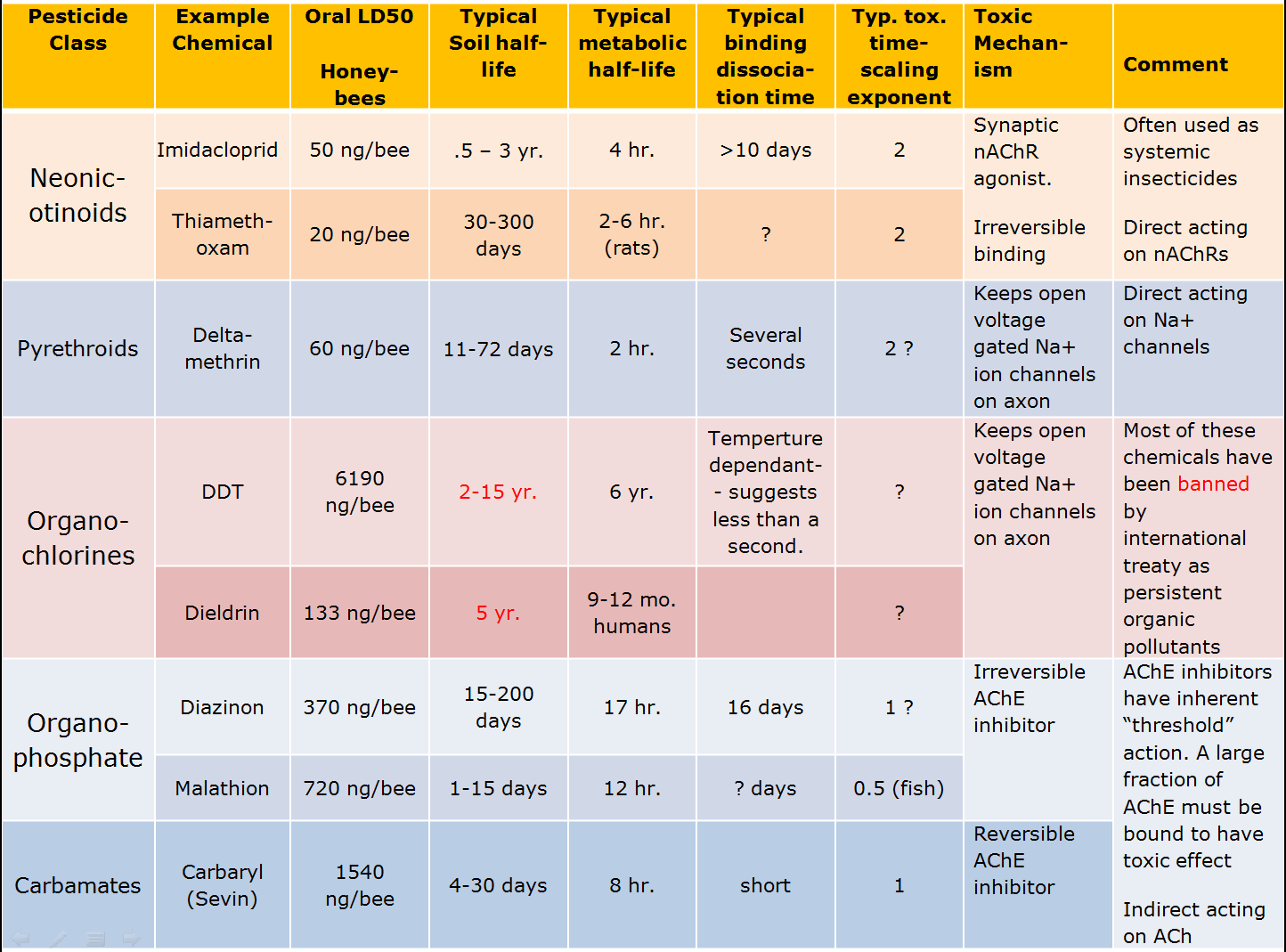
It is worth looking at the typical chemicals in the table above in light of the requirements we identified as desirable for a safe pesticide. Note that the organochlorines failed badly because they were so persistent in the environment to the point they have been almost universally banned. They were largely replaced by the organophosphates with which we’ve continue to have an uneasy coexistence for the last half-century. Under scrutiny because of their potent effects on humans and other vertebrates, many of the organophosphate insecticides are being forced into retirement. The replacement has been the neonicotinoids, which have the benefit of relative specificity to invertebrate nAChR receptors making the chemicals less toxic to humans and other vertebrates. Unfortunately, the neonicotinoids fail with regard to all three of the properties for safe and effective pesticides.
From our discussion you can see that the safest chemicals are the carbamates. Typically it takes more chemical (compare LD50 for neonicotinoids, organophosphates, and carbamates) to kill the target insect, but the persistence of the chemical in the environment is short. It is metabolized relatively quickly, and acts reversibly with the target receptors. Finally, it is also an AChE inhibitor that has a strong threshold of action effect. Compare this with the neonicotinoids at the top of the chart. It takes much less neonicotinoid chemical to kill, but this is likely due to its tenacious persistence on the target receptor sites. The chemicals do not degrade very quickly in the environment so they will continue to accumulate on target and non-target organism synaptic receptors long after the initial application. And finally, the neonicotinoids produce toxic effects at residual dose levels, unlike the AChE inhibitors. All of the tricks we have in the playbook to segregate between target and beneficial insects fail with the neonicotinoids.
Implications of threshold action for toxicity scaling
Change of sign of the acetylcholine growth rate provides a clear qualitative turning point for the organism. It is easy to understand how such a runaway event can lead to death. Hence, if you wish to model the toxicity scaling of a compound with such a distinct threshold action, all you have to do is follow the movement of toxin to receptor sites until the threshold is reached. This will naturally give you Haber’s rule for substances that accumulate, such as most of the organophosphate insecticides. For insecticides that don’t accumulate on receptors, such as carbamates, one would expect threshold action without a significant time dependence. Once pesticide concentrations reached levels where chemical equilibrium at receptor sites resulted it enough bound AChE to change the sign of the ACh growth rate, the threshold condition would be reached. However, at small residual concentrations of acetylcholinesterase inhibitors, the molecules disable a few AChE sites and hence slightly change the synaptic response, but otherwise remain largely benign to the organism. For this class molecules, there is a very large change in toxic effect with concentration. Despite the continued environmental issues and concerns with organophosphate pesticides, it should be recognized that they may be intrinsically environmentally safer because of their strong threshold action than the newer neonicotinoids.
For the neonicotinoids where there is no distinct threshold condition, the situation is more complicated. The transition from alive to dead is not accompanied by a convenient mathematical marker like the change in sign of a growth rate. Especially at the residual limit, we are left to speculate on the physiological impact of accumulate insults from the toxic chemical. Single molecules will open ion channels and begin to depolarize neurons. This abnormal state of affairs would be countered by energy-burning processes in the organism to mitigate the dysfunction. This is the definition of stress. It is likely that the residual-level stresses to non-target organisms is the Achilles’ heal for the neonicotinoid insecticides. Very low concentrations of these pesticides have the potential to switch on compensatory physiological processes that are poorly understood, but likely stressful. One example was the discovery that very low levels of the neonicotinoid clothianidin reduced the immune response of honeybees to the point where deformed wing virus could replicate. Low levels of the acetylcholinesterase inhibitor chlorpyriphos, the molecules of which in our understanding would be rather benignly latched on to a few of the AChE sites, showed no such immune suppression effect.(9) The fact that nAChR channels are involved in less well studied immune system and cellular signaling functions adds to the risk that disrupting these pathways will have unintended consequences.(10,11)
A key point is that at residual levels, AChE inhibitors are really doing nothing. A small fraction of the AChE sites my be out of commission, but even that effect is only apparent when the neuron fires and there is ACh to be swept away. During the neuron’s quiet state the pesticide molecules are benign. Contrast this situation with what happens on the postsynaptic membrane with a few neonicotinoid molecules. Single neonicotinoid molecules hold open nAChR channels that will tend to depolarize the neuron. This happens even when the neuron is in an un-stimulated state. However, given the persistent depolarization by the open channel, it can’t rest. Instead the cell must muster energetic processes in an attempt to restore the neuron’s polarization so that it may still function.
Besides suppression of immune response as mentioned above, there are likely other detrimental effects from the energy sapping response required by residual neonicotinoid poisoning. Trade-offs between energy expenditures to maintain neurological function and more normal activities such as powering flight muscles may explain some of the observed effects of chronic low level exposure. (12) Another study shows epi-genetic changes to imidacloprid-exposed honeybee larva that that strongly affects genes involving metabolism. (13) The myriad effects that low level neonicotinoid exposure presents, such as impaired navigation, poor learning ability, reduced flight time, and immunological impairment may be better understood from the perspective of the metabolic stress caused by open nAChR channels than by direct neurological impairment.
(1) Fetz EE, Gustafsson B. Relation between shapes of post-synaptic potentials and changes in firing probability of cat motoneurones. The Journal of Physiology. 1983;341:387-410.
(2) Bryan Ballantyne and Timothy C. Marrs., Clinical and Experimental Toxicology of Organophosphates and Carbamates, Published 1992 by Butterworth-Heinemann.
(3) Waseem M, Perry C, Bomann S, Pai M, Gernsheimer J. Cholinergic Crisis after Rodenticide Poisoning. Western Journal of Emergency Medicine. 2010;11(5):524-527.
(4) Gadsby DC. Ion channels versus ion pumps: the principal difference, in principle. Nature reviews Molecular cell biology. 2009;10(5):344-352.
(5) Rondeau G, Sánchez-Bayo F, Tennekes HA, Decourtye A, Ramírez-Romero R, Desneux N. 2014 Delayed and time-cumulative toxicity of imidacloprid in bees, ants and termites. Sci. Rep. 4.
(6) Main AR, Headley JV, Peru KM, Michel NL, Cessna AJ, Morrissey CA (2014) Widespread Use and Frequent Detection of Neonicotinoid Insecticides in Wetlands of Canada’s Prairie Pothole Region. PLoS ONE 9(3): e92821.
(7) Kathryn L. Klarich, Nicholas C. Pflug, Eden M. DeWald, Michelle L. Hladik, Dana W. Kolpin, David M. Cwiertny, and Gregory H. LeFevre, Occurrence of Neonicotinoid Insecticides in Finished Drinking Water and Fate during Drinking Water Treatment, Environmental Science & Technology Letters 2017 4 (5), 168-173.
(8) Sadaria AM, Supowit SD, Halden RU. Mass Balance Assessment for Six Neonicotinoid Insecticides During Conventional Wastewater and Wetland Treatment: Nationwide Reconnaissance in United States Wastewater. Environmental Science & Technology. 2016;50(12):6199-6206.
(9) Di Prisco, G. et al. Neonicotinoid clothianidin adversely affects insect immunity and promotes replication of a viral pathogen in honey bees. PNAS 110, 18466–18471, (2013).
(10) Zdanowski R, Krzyżowska M, Ujazdowska D, Lewicka A, Lewicki S. Role of α7 nicotinic receptor in the immune system and intracellular signaling pathways. Cent Eur J Immunol. 2015;40(3):373-9.
(11) Pamminger T, Botías C, Goulson D, Hughes W.O.H. A mechanistic framework to explain the immunosuppressive effects of neurotoxic pesticides on bees. Functional Ecology; 2018; 32(8):1921-30.
(12) Tosi S., Burgio G., Nieh J.C. A common neonicotinoid pesticide, thiamethoxam, impairs honey bee flight ability. (2017) Scientific Reports, 7 (1) , art. no. 1201.
(13) Derecka K., Blythe M.J., Malla S., Genereux D.P., Guffanti A., Pavan P., Moles A., Snart C., Ryder T., Ortori C.A., Barrett D.A., Schuster E. and Stöger R., 2013. Transient exposure to low levels of insecticide affects metabolic networks of honeybee larvae PLOS ONE. 8(7):e68191.
This article in PDF format here: Threshold mechanisms in acetylcholine pathway insecticides and environmental safety

Well, most of this is beyond me, but I think I understand enough to maybe solve my problem. 6 years ago I was living in ND among sugar beet growers who regularly used all sort of chemicals on their farms. I started having nerve problems, twitching, pain other things. It was so weird. Although I have gotten better since moving to the PNW, it hasn’t completely gone away and I have them occasionally. Also, I pick up ever sickness anyone near me has. I am wondering if I have something similar to the bees. Anyway, all the more reason to stay far away from all those chemicals and get my husband off his “perfect yard” kick. Thanks for your blog!
Hi Ann, I briefly looked at the insecticides that the sugar beet producers use, and it seems that Chlorpyrifos is a favorite. This is a powerful organophosphate insecticide that is banned many places because of its potential harm to people – usually farm workers and children. You may wish to read about the symptoms of poisoning from this chemical. If you were really “living among sugar beet growers” and were getting sick with nerve and twitchy pain this could be the culprit. You may also wish to talk to a toxicologist. Hopefully no longer being exposed will improve your situation.
This shows why the chemical companies want to switch over to the neonicoitinoids — because indeed the OP chemicals are very dangerous ones. The only problem is that the neonicotinoids are much worse for all invertebrates at low level chronic doses that the OP’s are. Best just to figure out how the organic growers do it and live without all of the chemicals!
Gary